Газовая хроматография
Газовая хроматография — один из многих видов хроматографии. Описанная впервые в 1952 г. она стала популярной благодаря:
• быстроте и легкости, с которой могут быть проанализированы сложные смеси;
• очень малой требуемой пробы для анализа;
• гибкости и надежности используемого оборудования.
Фазовые системы
Этот термин в газовой хроматографии обозначает комбинацию используемых подвижной и неподвижной фаз.
Неподвижная фаза состоит из твердых частиц, предпочтительно с узким интервалом по размерам. Их средний размер обычно 0,1-0,3 мм, хотя в некоторых случаях для достижения очень высокой эффективности газохроматогра-фических колонок используются частицы меньшего размера. С точки зрения химического состава и свойств используемые неподвижные фазы могут быть подразделены на три группы.
1) адсорбенты, обычно с очень большой удельной поверхностью (50-1000 м2/г): силикагель, оксид алюминия, молекулярные сита, активный уголь и графитированная сажа. Газоадсорбционная хроматография — не очень распространенный метод, за исключением анализа газов или решения особых задач;
2) нейтральные, или так называемые инертные носители, обычно получают из диатомитовых материалов, иногда из полимеров.
На них наносится жидкость с очень низким давлением пара и высокой термической стабильностью в условиях использования колонки. В настоящее время газожидкостная хроматография является самым распространенным методом.
В газовой хроматографии наиболее часто используются следующие инертные носители: карбопак (А, В, С), хромо-сорб (A, W, G, Р), молекулярные сита, графитированная сажа, цеолиты и др.
В качестве неподвижной фазы в газожидкостной хроматографии наиболее часто используются: апиезон М, карбо-вакс 20М, карбовакс 1500, дексил 300, дексил 400, дибутил-фталат, диэтилен-гликольадипат, динонилфталат, полифе-ниловый эфир, полипропиленгликоль, поливинилпирролидон, силикон GESF 96, силикон GEXE 60, силикон SE 30, фенил-силикон SE 52 и др.;
3)для проведения очень трудных разделений успешно используются также адсорбенты с нанесенным на них малым количеством жидкости с низким давлением пара. Этот метод обычно называется газовой хроматографией на адсорб
ционных слоях или газоадсорбционной хроматографией на модифицированных адсорбентах.
Подвижной фазой служит инертный газ (гелий, азот, аргон) или такой газ, как водород, который в условиях газовой хроматографии проявляет себя как инертный. В некоторых случаях используют водяной пар или безводный аммиак. Химический состав газа-носителя оказывает весьма незначительное влияние на удерживание веществ и на их разделение.
С другой стороны, физические свойства подвижной фазы и особенно значительная сжимаемость газов, большое значение коэффициента диффузии и большое различие между парциальными молярными объемами в подвижной и неподвижной фазах имеют существенное влияние и являются причиной значительных различий между газовой хроматографией и жидкостной.
Схематическое описание газового хроматографа
Имеется много практических реализаций принципов газовой хроматографии, однако по своим основным конструктивным особенностям вся ГХ-аппаратура очень схода. На рис. 4.5 приведено схематическое описание газового хроматографа.
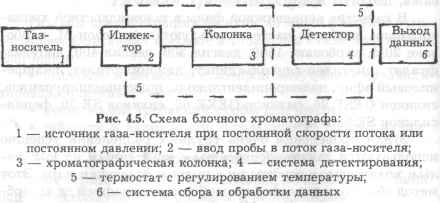
Основными блоками газового хроматоргафа являются следующие:
• Блок подготовки газа-носителя, который подает стационарный поток выбранного газа-носителя. В самых распространенных системах используется регулятор скорости потока. Массовая скорость потока газа-носителя через этот регулятор поддерживается постоянной. Другими словами, число молей газа, проходящего через колонку в единицу времени, является постоянным.
• Система ввода проб, которая обеспечивает ввод точного количества пробы в этот поток газа точно в начало колонки. Эта проба должна испаряться за достаточно короткое время и вводиться в колонку в виде цилиндрической пробки пара, разбавленного газом-носителем.
• Колонка, которая установлена в термостате с регулированием температуры. Выбираемая температура обычно заключается в диапазоне от комнатной температуры до 350°С, хотя были описаны анализы в более широком диапазоне (от -180 С до +1000°С).
• Детектор, который обеспечивает сигнал, пропорциональный составу газа-носителя. Желательно, чтобы этот сигнал был нулевым, когда из колонки выходит чистый газ-носитель, и пропорциональным концентрации любого вещества, отличающегося от газа-носителя. Такой детектор называется линейным. Если коэффициент пропорциональности одинаков для всех веществ, детектор называется идеальным. На практике идеальный детектор не существует.
Компоненты смеси переносятся по колонке газом-носителем. Они движутся со скоростью, которая пропорциональна линейной скорости газа-носителя, но меньше ее и зависит от силы взаимодействия каждого из этих компонентов с неподвижной фазой.
Соответственно, если неподвижная фаза была выбрана правильно, каждый компонент находится в колонке или элюируется разное время и отделяется от других компонентов. Сигнал детектора позволяет проводить идентификацию каждого компонента по времени элюирования его зоны (также называемом его временем удерживания) и его количественное определение по величине сигнала детектора (его высоте или площади).
Таким образом, хроматографический процесс является последовательным процессом. Каждому вводу пробы соответствует разделение, сопровождающееся детектированием.
Экспериментальные хроматографическиеданные
Из данных, записываемых во время хроматографического анализа, можно отметить пять параметров для каждого пика, допуская, что он довольно хорошо отделен от соседнихпиков (рис. 4.6).
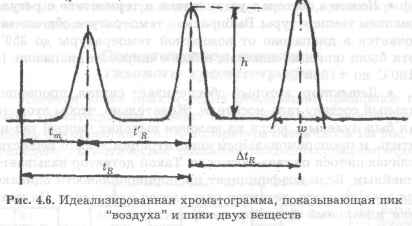
Этими пятью основными экспериментальными данными являются: время удерживания, время задержки газа, ширина пика, высота пика, площадь пика.
1. Время удерживания tR.
Это время между вводом пробы и появлением на выходе из колонки максимальной концентрации зоны соответствующего вещества. По времени удерживания проводится идентификация разделяемых веществ.
2. Время задержки газа tm. Это время удержания инертного вещества, которое не удерживается на колонке, т. е. вещества, не адсорбируемого или не растворяемого неподвижной фазой.
3. Ширина пика w. Ширина пика обычно определяется как длина сегмента нулевой линии, измеряемая между точками пересечения с нулевой линией двух касательных в точках перегиба пика. Используется также ширина пика на половине его высоты или на некоторой другой промежуточной высоте.
4. Высота пика h.
Это расстояние между нулевой линией и максимумом пика.
5. Площадь пика А, которая в настоящее время измеряется интегрированием сигнала. По площади пиков проводится количественный анализ.